
1Žaneta Grácová, 1Miriam Malá, 1Alena Holčíková, 1Barbora Jeřábková, 2Renata Gaillyová, 3Hana Vinohradská, 1Lukáš Homola
1 Klinika dětských infekčních nemocí, Fakultní nemocnice Brno
2 Ústav lékařské genetiky a genomiky, Fakultní nemocnice Brno
3 Oddělení klinické biochemie, Fakultní nemocnice Brno
Souhrn
Cystická fibróza (CF) je závažné nevyléčitelné autozomálně recesivně (AR) podmíněné genetické onemocnění způsobené mutací genu CFTR (cystic fibrosis transmembrane conductance regulator). Dochází při ní k poruše transportu chloridových
a hydrogenuhličitanových iontů přes membránu buněk a následnému zahuštění tělesných sekretů. Následkem stázy hustého hlenu dochází k postižení a postupnému selhání orgánů.
U většiny pacientů se choroba manifestuje pod obrazem chronického sinobronchiálního onemocnění, pankreatické insuficience a zvýšené koncentrace chloridů v potu.
Novorozenecký screening (NS) je celoplošné aktivní vyšetření novorozenců, jehož cílem je zachytit nejčastější vrozené vady. Od roku 2022 se z NS vyšetřuje 20 nemocí včetně CF, která byla do programu zařazena v roce 2009. V české republice (ČR) spočívá NS ve stanovení imunoreaktivního trypsinogenu (IRT) a genetického vyšetření [1]. Výsledek screeningu může být negativní, tedy CF je nepravděpodobná, nebo pozitivní. Děti s pozitivním výsledkem screeningu jsou odeslány do CF centra FN Motol (z Čech) nebo FN Brno (z Moravy), kde následně proběhne další vyšetření, potní test a podle výsledku konzultace, případně hospitalizace s edukací.
Klíčová slova
cystická fibróza, CF SPID, novorozenecký screening, imunoreaktivní trypsinogen, potní test
Summary
Cystic fibrosis (CF) is a severe incurable autosomal recessive (AR) genetic disease caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene. It results in impaired transport of chloride and bicarbonate ions across the cell membrane and subsequent thickening of body secretions. Stasis of thick mucus results in organ involvement and failure. In the majority of patients, the disease manifests itself under the image of chronic sinobronchial disease, pancreatic insufficiency and increased chloride concentration in sweat.
Newborn screening (NS) is a full-body active examination of newborns aimed at catching the most common birth defects. As of 2022, NS screens for 20 diseases, including CF, which was added to the program in 2009. In the Czech Republic, NS consists of IRT (immunoreactive trypsinogen) and genetic testing. [1] The screening result may be negative and therefore CF is unlikely or positive. Children with a positive screening result are referred to the CF centre of the Motol University Hospital, Brno, where further examination, sweat test and, depending on the result, consultation or hospitalization with education are performed.
Keywords
cystic fibrosis, CF SPID, newborn screening, immunoreactive trypsinogen, sweat test
Vyšetření suché kapky krve
V rámci novorozeneckého screeningu CF (NSCF) se všem novorozencům již v porodnici odebírá suchá kapka krve z patičky mezi 48. a 72. hodinou po narození. V současné době probíhá v ČR třístupňový model – IRT/DNA/IRT.
V první fázi je stanovena koncentrace IRT. Cut off hodnota činí 65 ng/ml. Toto jednorázové vyšetření není specifické, mnoho zdravých novorozenců může mít přechodně zvýšenou hodnotu IRT, proto se všem novorozencům s hypertrypsinogenemií provádí genetická analýza 50 nejčastějších mutací genu CFTR [2]. Bylo nalezeno více než 2 000 mutací tohoto genu [3] a z těchto 2 000 mutací je 401 spojeno s obrazem CF [4]. Nejčastější mutací asociovanou s CF je mutace F508del [5]. Incidence tohoto geneticky podmíněného onemocnění je 1 : 4 500 a každý 42. je přenašečem CF [1]. DNA analýza se provádí ze suché kapky, tudíž se eliminuje nepříjemný krevní odběr u čerstvě narozených novorozenců. Jestliže se nalezne 1 nebo 2 mutace, následuje provedení potního testu. Při záchytu vysokého IRT bez záchytu patogenní mutace CFTR, se opakuje test suché kapky krve ke stanovení IRT, tzv. IRT recall a v případě pozitivity je nutné provést potní test [6].
Za posledních 9 let, kdy se provádí novorozenecký screening, se zachytilo celkem 160 případů CF; 121 pacientů bylo pozitivních z NS a celkem bylo vyšetřeno 1 037 308 novorozenců, 59 CF SPID (CF Screen Positive Inconclusive Diagnosis). V roce 2022 bylo z novorozeneckého screeningu diagnostikováno celkem 17 novorozenců s CF [7].
V rámci NSCF se suchá kapka odebraná z patičky novorozence odesílá z celé Moravy a Slezska do FN Brno. V případě záchytu zvýšené hodnoty IRT existuje úzká spolupráce biochemika, genetika a lékaře CF centra. O rizikových novorozencích je vedena databáze. Pediatr a rodiče těchto rizikových dětí (tzn. těch, které mají zvýšenou hodnotu IRT a záchyt patogenní mutace CF) obdrží z CF centra zvací dopis. Úkolem pediatra je informovat rodiče, že u jejich dítěte vyšel pozitivní screening pro CF, který ještě neznamená, že dítě trpí CF, ale je nutné provést další vyšetření, především potní test a klinické vyšetření k potvrzení, či vyloučení diagnózy.
Potní test (PT)
PT představuje vyšetření chloridů v potu. Děti s CF se také označují jako slané děti, neboť více ztrácejí sůl, a jejich kůže je tedy slanější. Tento test je bezbolestný, provádí se ambulantně, nevyžaduje žádnou přípravu a trvá okolo 30 minut. Pot se odebírá z předloktí, kde dochází ke stimulaci potních žláz pilokarpinovou iontoforézou. Normální koncentrace chloridů v potu je do 30 mmol/l, hodnoty 60 mmol/l a vyšší svědčí pro diagnózu CF. Hodnoty mezi 30 a 59 mmol/l potu jsou tzv. hraniční. PT se provádí u dětí po 2. týdnu života, jejichž váha je větší než 3 000 g. U menších dětí či nedonošených dětí hrozí riziko odběru nedostatečného množství potu k analýze, a je tedy nutné test opakovat. Zisk dostatečného množství potu je podmínkou správného vyšetření (> 100 mg při sběru do filtračního papíru nebo > 15 µl při sběru do Macroduct) [8] (tabulka č. 1).
Stanovení diagnózy
Po provedení všech vyšetření mohou nastat následující situace:
Negativní potní test, nalezena 1 patologická mutace CFTR
I v tomto případě jsou rodiče s dítětem pozváni do centra pro léčbu CF, jsou informováni o všech výsledcích. Lékař odebere adekvátně anamnézu zaměřenou na rizikové aspekty týkající se CF, např. porodní váha, týdenní přírůstky váhy, charakter stolice, steatorea, a následně je dítě klinicky vyšetřeno. Novorozenec je v tomto případě zdravým přenašečem onemocnění CF a rodina je informována o přenašečství, riziku přenosu patologické mutace na další generace a možnostech genetického vyšetření budoucího partnera dítěte a rodiny. Další dispenzarizace v centru CF neprobíhá.
Pozitivní potní test a 1 nebo 2 pozitivní mutace genu CFTR
Dítě má CF a následuje edukace rodiny za hospitalizace v CF centru (FN Motol, FN Brno). Během hospitalizace se provádějí další vyšetření, do kterých patří krevní odběry, konfirmace genetického vyšetření ze žilního odběru, zobrazovací metody (UZ břicha, rtg. S + P), kardiologie, vyšetření odpadů v moči a podle výsledků se zahajuje symptomatická léčba (inhalace hypertonické soli, dechová rehabilitace, pankreatická substituce, substituce vitamínů rozpustných v tucích, suplementace soli). Péče o pacienty s CF je multidisciplinární a další odborníci, jako např. sociální a rehabilitační pracovnice, nutriční terapeuti, psychologové, jsou zapojeni do edukace a péče o celou rodinu. Pokud byla screeningem nalezena pouze jedna causing mutace pro CFTR, je nutné provést sekvenaci DNA k nalezení méně časté, tedy vzácnější mutace CF.
CF SPID (CF screening positive inconclusive diagnosis)
Do této skupiny zařazujeme pacienty, kteří jsou nosiči jedné (nebo žádné) mutace CFTR a mají hraniční potní test 30–59 mmol/l. Nebo jsou nosiči dvou mutací CFTR, kde minimálně jedna je s nejasným významem, a mají negativní potní test, tedy chloridy v potu do 30 mmol/l. CF SPID obvykle nemá žádné příznaky, nicméně je potřeba tyto děti sledovat v centru CF, protože podle recentních studií cca 10 % CF SPID bylo v průběhu následujících let překlasifikováno na CF [9] (tabulka č. 2).
Závěr
NSCF je v ČR na kvalitní úrovni. U CF dochází k postupné manifestaci klinických příznaků a progresi klinického stavu, tudíž brzký záchyt nemoci je esenciální. Jestliže je léčba zahájena včas, ještě před manifestací klinických příznaků, výrazně se zlepšuje celková prognóza onemocnění i kvalita života pacienta. V současné době existuje na trhu velice efektivní terapie modulátory CFTR, která koriguje nefunkční protein CFTR. Některé tyto léky se mohou pacientovi nasadit již v brzkém věku života, a tedy i proto je včasná diagnóza onemocnění velice důležitá.
Graf č. 1 Stanovení CF
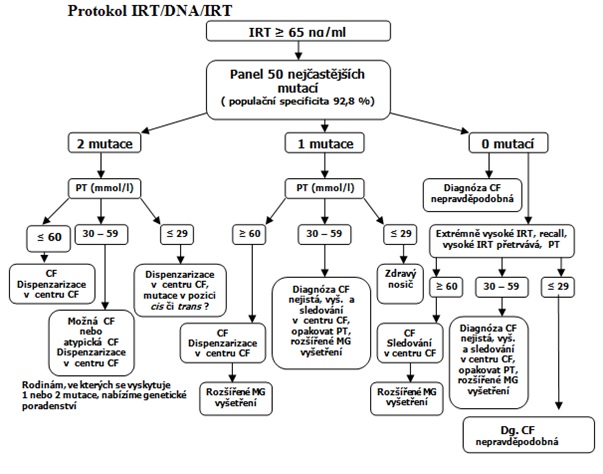
Zdroj: SOP pracovní skupina Pracovní skupina pro cystickou fibrózu ČLS JEP. SOP stanovení diagnózy CF 2017 [online]. Dostupné na http://www.pracovniskupinacf.blogspot.com
Tabulka č. 1 Hodnoty potního testu
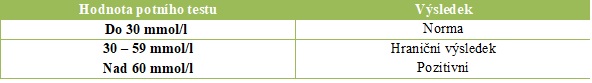
Zdroj: archiv autorů
Tabulka č. 2 Kritéria diagnózy CF SPID

Zdroj: archiv autorů
Použité zdroje:
Novorozenecký screening. Cystická fibróza [online]. [cit. 2023-12-20]. Dostupné z: https://www.novorozeneckyscreening.cz/
Castellani C, Tamani A, Mastella G. Protracted neonatal hypertrypsinogenaemia, normal sweat chloride, and cystic fibrosis. ARch Dis Child. 2000; 82: 481–482.
Cystic fibrosis mutation database (CFMDB) [online], 2022. Dostupné na: http://www.genet.sickkids.on.ca/StatisticsPage.html
CFTR2 database [online], 2022. Dostupné na: https://cftr2.org/mutations_history
Cystic fibrosis foundation (CFF) [online], 2022. Dostupné na: https://www.cff.org/research-clinical-trials/types-cftr-mutations
SOP Pracovní skupina pro cystickou fibrózu ČLS JEP. SOP genetické poradenství a testování u CF [online]. Dostupné na http://www.pracovniskupinacf.blogspot.com
Bartošová J. Novorozenecký screening cystické fibrózy a diagnostika CFSPID. Čs Pediat. 2019; 74 (7): 381–386.
SOP pracovní skupina Pracovní skupina pro cystickou fibrózu ČLS JEP. SOP stanovení diagnózy CF [online], 2017. Dostupné na http://www.pracovniskupinacf.blogspot.com
SOP pracovní skupina Pracovní skupina pro cystickou fibrózu ČLS JEP. SOP CF SPID [online], 2017. Dostupné na http://www.pracovniskupinacf.blogspot.com




